Rapidly Realizing PracticalApplications of Cutting-edge Technologies
2024.04.22
Engineering
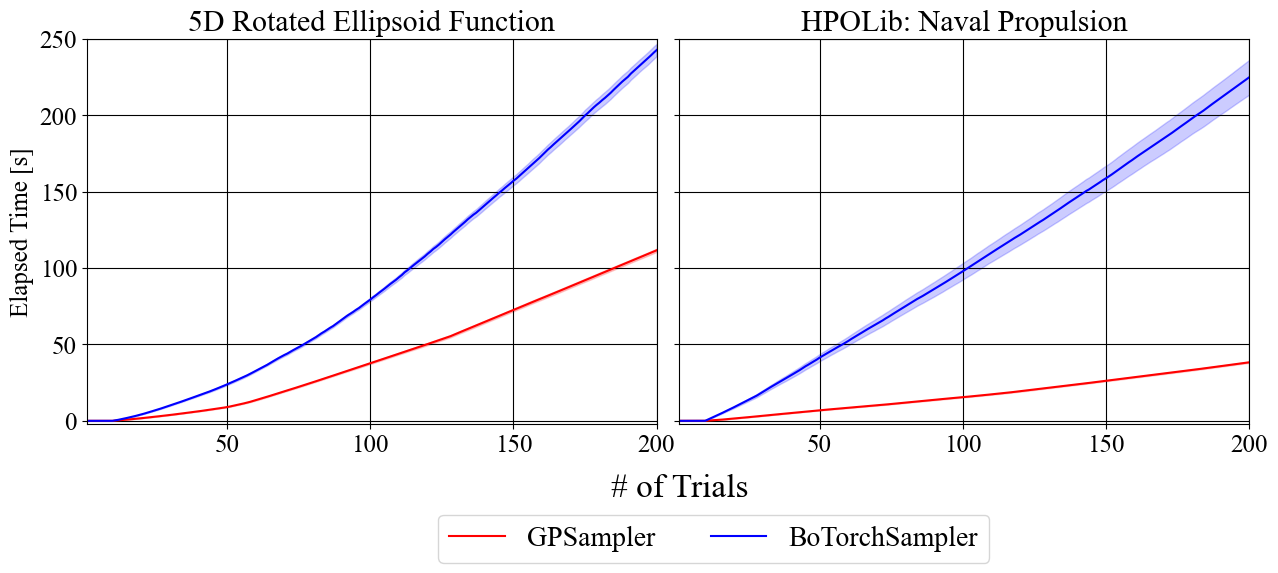
BoTorchに依存しないGPSamplerの導入
By : Shuhei Watanabe
2024.04.17
Research
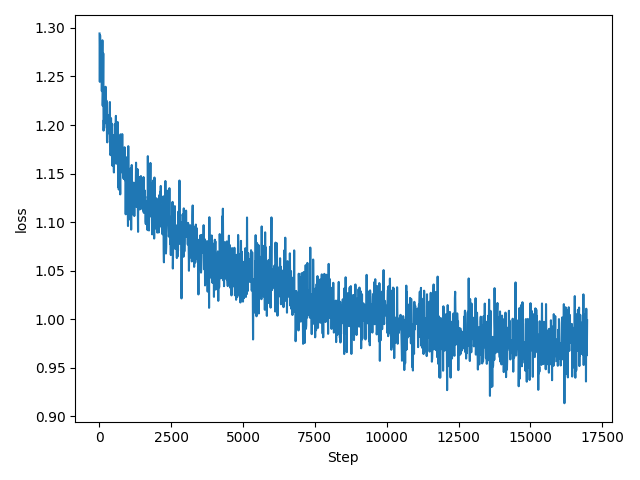
継続事前学習による金融ドメイン特化LLMの構築の検証
By : Masanori Hirano
2024.04.16
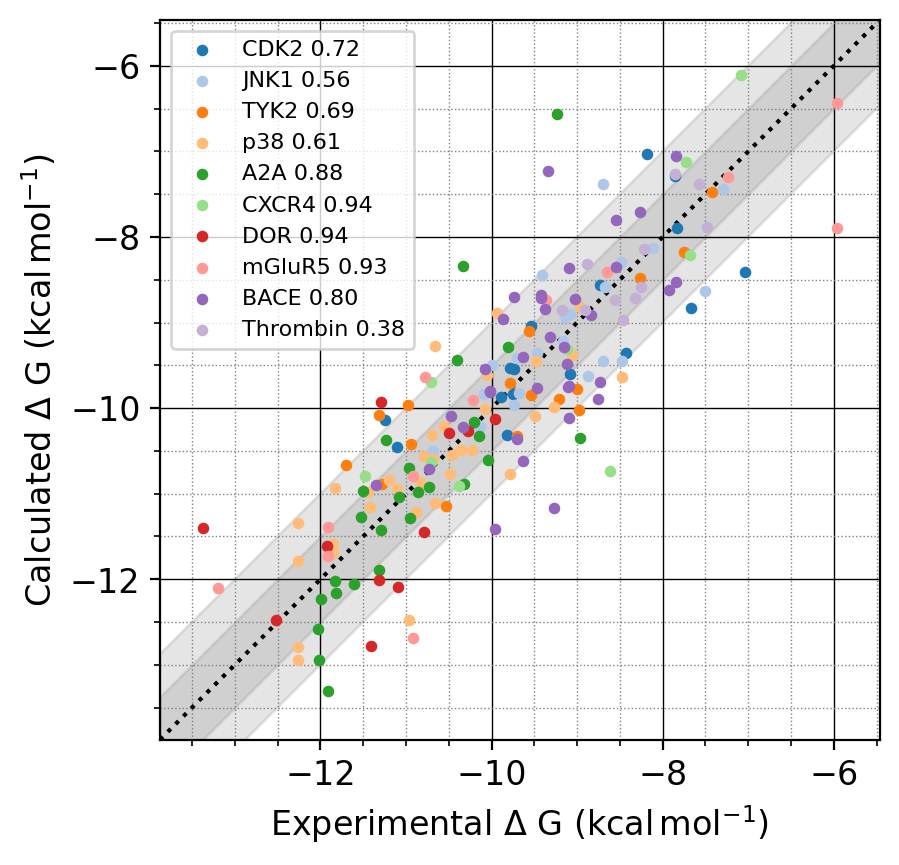
P-FEP (RBFEP計算サービス) 提供開始
By : Junya Yamagishi
2024.04.08
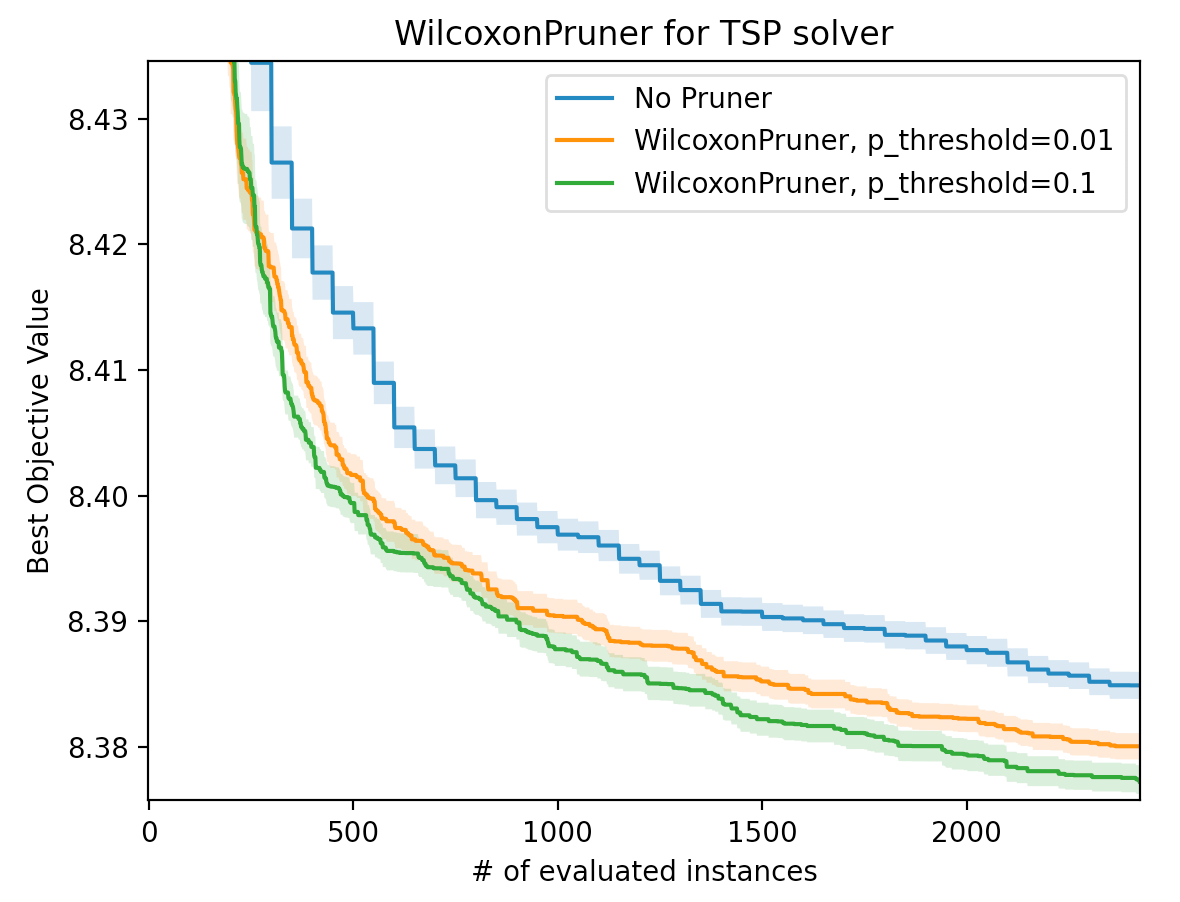
WilcoxonPruner: 統計的検定に基づくOptunaの新しい枝刈り
By : 王 允卓
2024.03.22
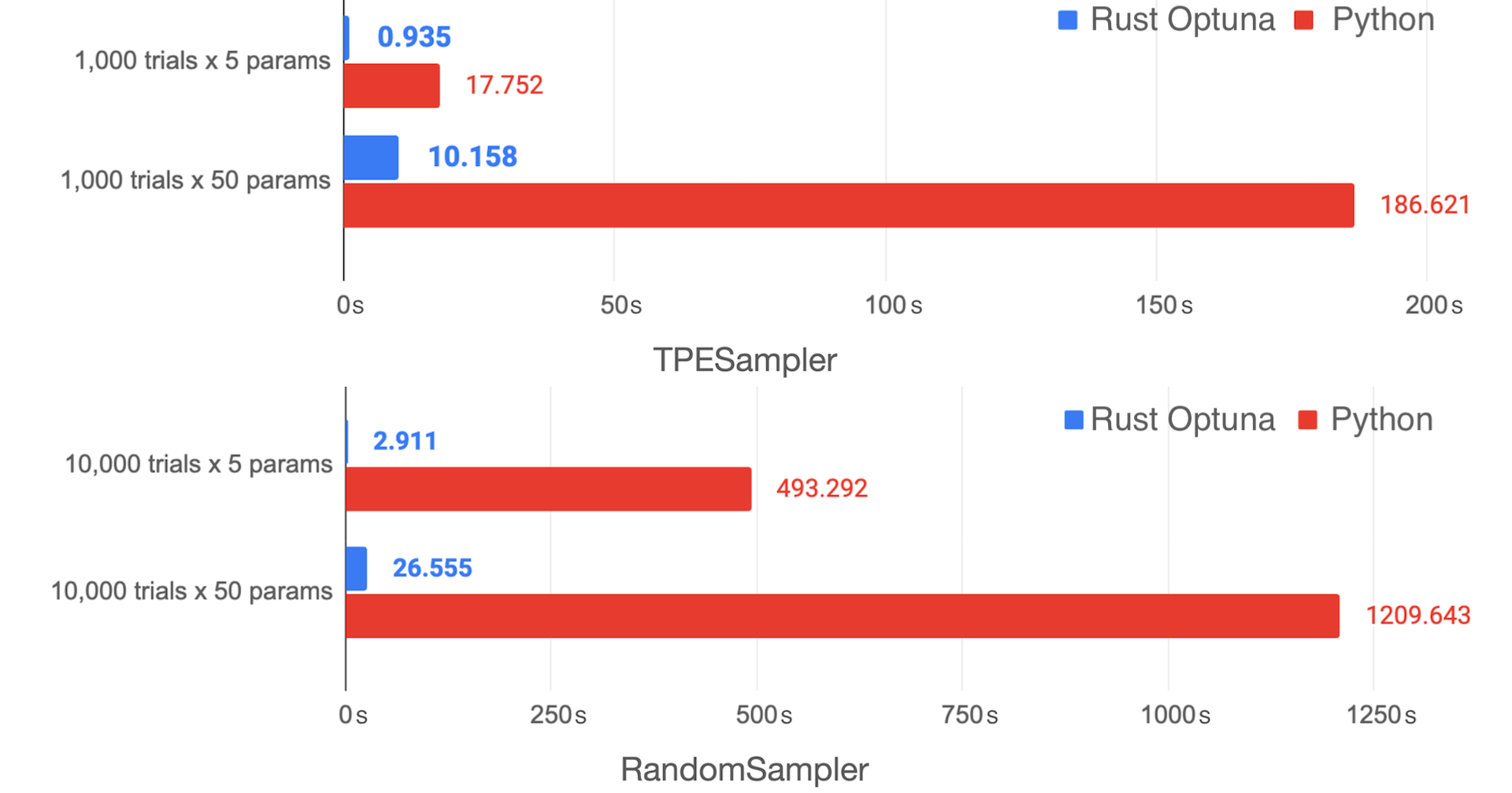
Rustによる高速なOptuna実装の試作
By : Masashi Shibata
2024.01.30
Large Language Models for Code (Code LLMs)と自然言語推論、ソースコードの関係について
By : Katsuhiko Ishiguro
2023.12.27
MN-Core におけるテンソルのメモリ配置レイアウト表現
By : Seiya Tokui
2024.03.18
Announcing Optuna 3.6
By : Hideaki Imamura
2023.12.19
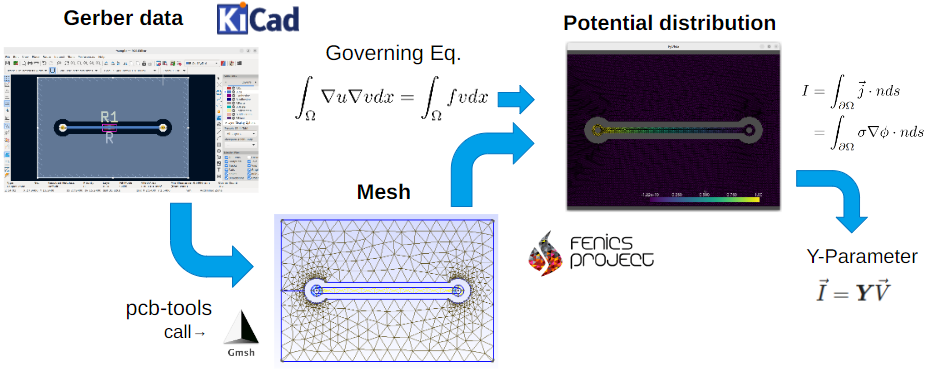
OSSによるプリント基板静電流シミュレーション
By : Takuya Yamauchi