Blog
* English blog is also written here.
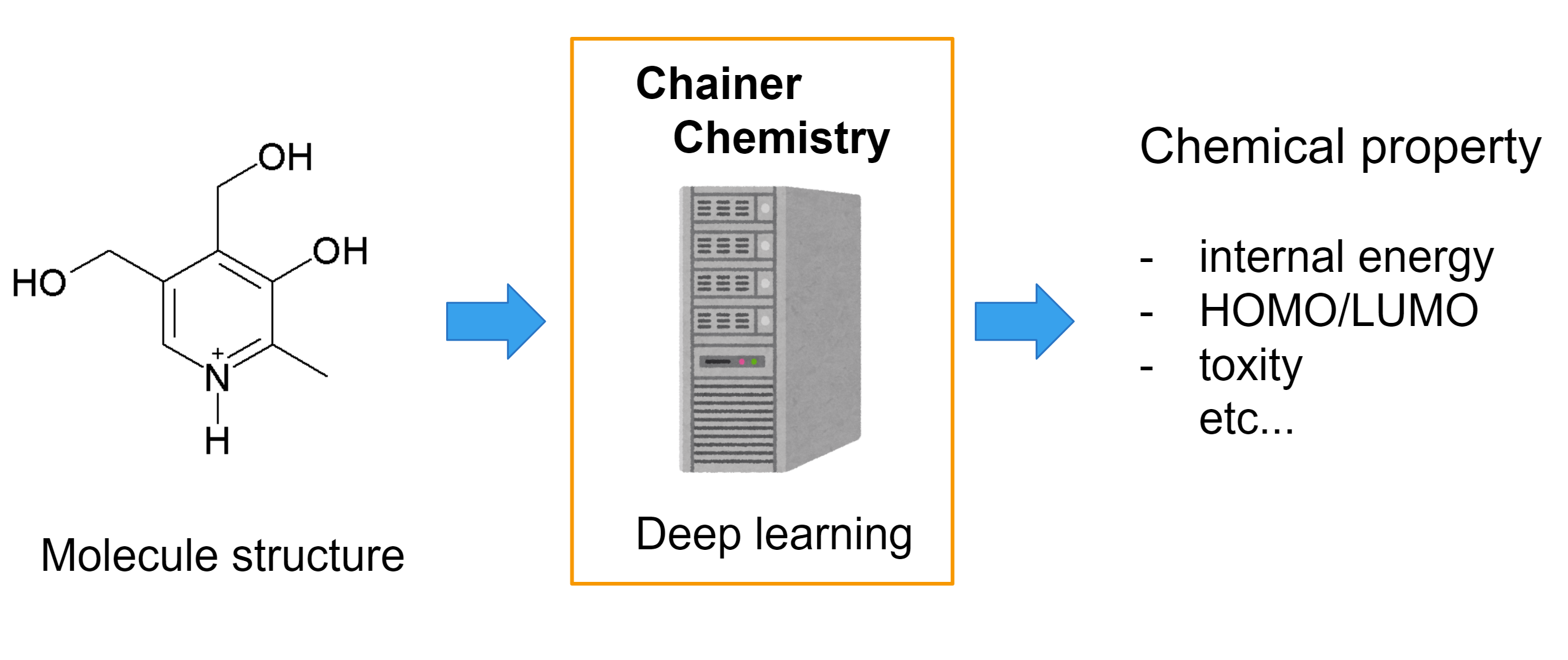
Chainer [1]を使った、化学、生物学分野のための深層学習ライブラリ Chainer Chemistry を公開しました。
- Github page: https://github.com/pfnet-research/chainer-chemistry
- Documentation: https://chainer-chemistry.readthedocs.io
本ライブラリにより、分子構造に対して簡単に深層学習(Deep learning)を適用することができるようになります。
例えば、化合物の分子構造を入力とした毒性の予測や、HOMO(最高被占軌道)レベルの回帰予測など、様々な化学的性質の予測に深層学習を適用することができます。
なお本ライブラリの開発にあたっては、PFN2017夏インターンシップに参加した京都大学の秋田大空さんにも実装に携わっていただきました。
特長
様々なGraph Convolutional Neural Network のサポート
Graph Convolutional Network (詳しくは下記参照)の登場により、”グラフ構造”を入力として深層学習が適用できるようになりました。Graph Convolutional Networkは現在盛んに研究がおこなわれていますが、本ライブラリでは今年発表されたばかりの論文も含めいくつかのネットワークを追実装しています。
現時点では以下のモデルが実装されています。
- NFP: Neural Fingerprint [2, 3]
- GGNN: Gated-Graph Neural Network [4, 3]
- WeaveNet: Molecular Graph Convolutions [5, 3]
- SchNet: A continuous-filter convolutional Neural Network [6]
データの前処理部分をライブラリ化・研究用データセットのサポート
様々なデータセットを共通のインターフェースで使えるように、ソフトウェアを設計しています。また、研究用によく使用されるデータセットに関してはライブラリ内でダウンロード・前処理を行うことができます。
現時点では以下のデータセットをサポートしています。
- QM9 [7, 8]: 9個までのC、O、N、F原子とH原子から構成された有機分子に対して、B3LYP/6-31GレベルのDFT(密度汎関数法)で算出されたHOMO/LUMOレベル、内部エネルギーなどの物性値をまとめたデータセット
- Tox21 [9]: 12種類のアッセイに対する毒性をまとめたデータセット
学習・推論コードのExample code を提供
ライブラリの使い方がわかるよう、モデルの学習コード・推論コードのExampleも公開しています。すでに実装済みのモデル・データセットに対して手軽に訓練・推論を試してみることができます。
背景
材料探索・創薬などの応用分野では、分子構造を入力とするシミュレーションが重要な位置を占めます。中でも量子力学的効果を高い精度で取り込みたい場合に用いられるDFT(密度汎関数法)のようなシミュレーション手法は、特に大きな分子に対して、膨大な計算量を要求することが知られています。このため、有用な新物質の候補となる多数の分子構造に対してシミュレーションを行うのが困難です。
そこで機械学習分野では、これまでに実測・計算されたデータを学習することにより、未知の分子の物性値を予測するというアプローチでの研究がおこなわれています。ニューラルネットワークを用いることにより、量子シミュレーションよりも高速に物性値の予測ができることが期待されています。
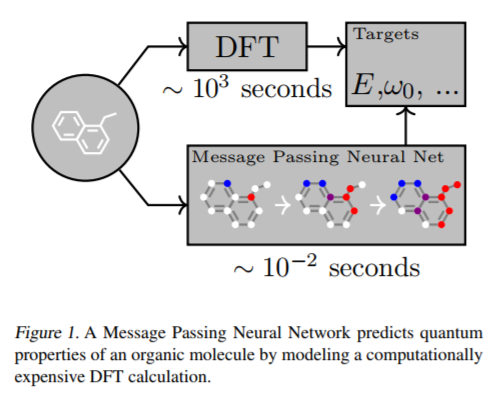
Cite from “Neural Message Passing for Quantum Chemistry”, Justin et al. https://arxiv.org/pdf/1704.01212.pdf
化合物に対して深層学習を適用することを考えた場合、その入出力をどのように扱うかということが問題となります。これは、通常の深層学習手法が固定長のベクトル値データを入力とするのに対し、分子構造は可変長で分岐やループを持ちうるデータ形式、つまりグラフであるためです。しかし、近年グラフ構造を扱うことのできるGraph Convolutional Neural Network が提案され、注目を集めています。
Graph Convolutional Neural Network とは
Convolutional Neural Network (畳み込みニューラルネットワーク)は、局所的な情報のみで計算を進める畳み込み層の導入によって、画像分類、セグメンテーションや画像生成などの分野で成功をおさめました。
Graph Convolutional Neural Network では、同様にグラフ上で近いノードに対する畳み込み演算を導入することにより、グラフ構造の取り扱いを可能にしています。
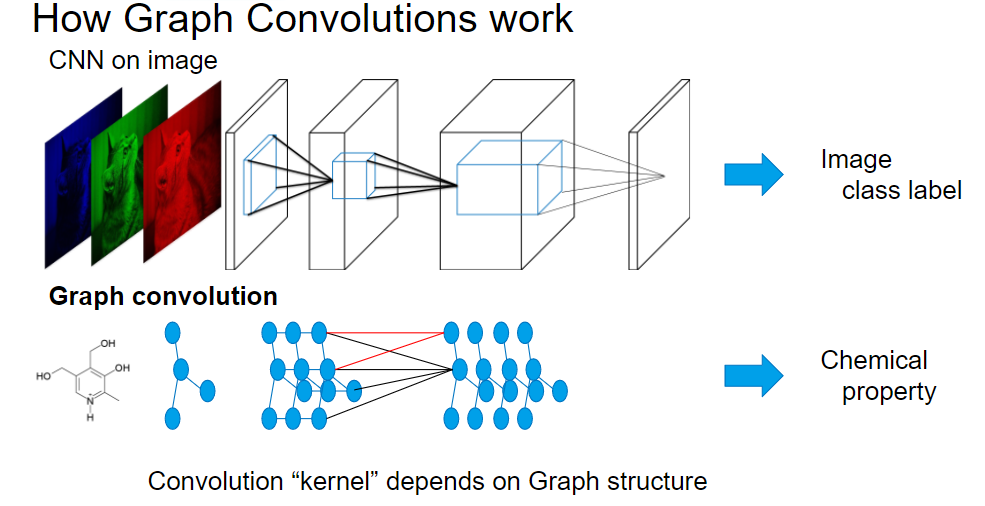
CNNが画像を入力とするのに対し、Graph CNNではグラフ構造(分子構造など)を入力として深層学習を行います。
グラフ構造を入力とするGraph Convolutional Neural Network は、分子構造にかぎらず、ソーシャルネットワークや交通網などに広く適用でき、ここ最近研究が進んできています。例えば、文献[10] では画像、[11]ではナレッジベース、[12]では交通量予測にGraph Convolutionを適用しています。
Graph Convolutional Networkに関しては、以下のブログでもわかりやすく説明されています。
対象ユーザー
- Deep learningの研究者
本ライブラリでは、最新のGraph Convolutional Neural Network の追実装を行っています。
今後、Graph Convolutionは計算科学に限らず、様々な分野への適用が考えられる技術なので、ぜひ様々な方に使っていただきたいです。 - 物質探索・創薬などの研究者
本ライブラリを用いることにより、様々な化合物に対する物性値予測のモデルを構築することができます。
今後の予定
本ライブラリはまだベータ版で、開発を行っているところです。今後は以下のような機能をサポートしていきたいと検討しています。
- Pre-trained modelを提供し、推論だけでの使用をサポート。
- データセットの拡充
- モデルの追加
Tutorial も用意しているので、ぜひ試してみてフィードバックをいただけるとありがたいです。
参考文献
[1] Tokui, S., Oono, K., Hido, S., & Clayton, J. (2015). Chainer: a next-generation open source framework for deep learning. In Proceedings of workshop on machine learning systems (LearningSys) in the twenty-ninth annual conference on neural information processing systems (NIPS) (Vol. 5).
[2] Duvenaud, D. K., Maclaurin, D., Iparraguirre, J., Bombarell, R., Hirzel, T., Aspuru-Guzik, A., & Adams, R. P. (2015). Convolutional networks on graphs for learning molecular fingerprints. In Advances in neural information processing systems (pp. 2224-2232).
[3] Gilmer, J., Schoenholz, S. S., Riley, P. F., Vinyals, O., & Dahl, G. E. (2017). Neural message passing for quantum chemistry. arXiv preprint arXiv:1704.01212.
[4] Li, Y., Tarlow, D., Brockschmidt, M., & Zemel, R. (2015). Gated graph sequence neural networks. arXiv preprint arXiv:1511.05493.
[5] Kearnes, S., McCloskey, K., Berndl, M., Pande, V., & Riley, P. (2016). Molecular graph convolutions: moving beyond fingerprints. Journal of computer-aided molecular design, 30(8), 595-608.
[6] Kristof T. Schütt, Pieter-Jan Kindermans, Huziel E. Sauceda, Stefan Chmiela, Alexandre Tkatchenko, Klaus-Robert Müller (2017). SchNet: A continuous-filter convolutional neural network for modeling quantum interactions. arXiv preprint arXiv:1706.08566
[7] L. Ruddigkeit, R. van Deursen, L. C. Blum, J.-L. Reymond, Enumeration of 166 billion organic small molecules in the chemical universe database GDB-17, J. Chem. Inf. Model. 52, 2864–2875, 2012.
[8] R. Ramakrishnan, P. O. Dral, M. Rupp, O. A. von Lilienfeld, Quantum chemistry structures and properties of 134 kilo molecules, Scientific Data 1, 140022, 2014.
[9] Huang R, Xia M, Nguyen D-T, Zhao T, Sakamuru S, Zhao J, Shahane SA, Rossoshek A and Simeonov A (2016) Tox21 Challenge to Build Predictive Models of Nuclear Receptor and Stress Response Pathways as Mediated by Exposure to Environmental Chemicals and Drugs. Front. Environ. Sci. 3:85. doi: 10.3389/fenvs.2015.00085
[10] Michaël Defferrard, Xavier Bresson, Pierre Vandergheynst (2016), Convolutional Neural Networks on Graphs with Fast Localized Spectral Filtering, NIPS 2016.
[11] Michael Schlichtkrull, Thomas N. Kipf, Peter Bloem, Rianne van den Berg, Ivan Titov, Max Welling (2017) Modeling Relational Data with Graph Convolutional Networks. arXiv preprint arXiv: 1703.06103
[12] Yaguang Li, Rose Yu, Cyrus Shahabi, Yan Liu (2017) Diffusion Convolutional Recurrent Neural Network: Data-Driven Traffic Forecasting. arXiv preprint arXiv: 1707.01926